Comprendre l’évolution des partenariats avec les organisations de patients
Chez Ipsen, nous menons chaque année une enquête afin de recueillir les retours des organisations de patients avec lesquelles nous collaborons à l’échelle mondiale. Cette démarche nous permet d’évaluer nos interactions, de mesurer la satisfaction et d’identifier des opportunités pour renforcer nos partenariats.
Les résultats de cette enquête apportent des enseignements précieux sur la manière dont notre approche évolue et sur la façon dont nous pouvons continuer à progresser.
En partageant ces analyses, nous contribuons à une meilleure compréhension des attentes des organisations de patients et à l’amélioration continue de nos pratiques.
Des bases solides fondées sur la confiance
Les organisations de patients évaluent généralement de manière positive nos interactions, notamment en matière de transparence, d’écoute et d’engagement.
Le dialogue ouvert et constructif reste au cœur de notre collaboration, permettant de bâtir des relations durables et significatives.
Dans le même temps, ces retours soulignent l’importance de maintenir des standards élevés et d’aller plus loin pour répondre aux attentes croissantes.
« À l’IKCC, notre partenariat avec Ipsen est ancré dans un engagement mutuel visant à valoriser la voix des patients et à favoriser une prise de décision partagée en oncologie. Ipsen démontre de manière constante un véritable engagement en faveur de la défense des intérêts des patients — que ce soit à travers des initiatives de formation, des publications, des événements collaboratifs ou des efforts continus de co-création. Ces actions durables garantissent que notre travail commun génère un impact social réel et joue un rôle essentiel dans la création de bénéfices durables pour les patients atteints d’un cancer du rein. »
Directeur exécutif, International Kidney Cancer Coalition (IKCC)
Un engagement solide, mais des attentes en hausse
L’enquête indique un niveau élevé de satisfaction en matière d’engagement, reflété par une note moyenne de 8,8 sur 10.
Ce résultat démontre que les efforts d’Ipsen pour construire des relations constructives sont reconnus par les organisations de patients. Cependant, il met également en évidence des attentes croissantes. Ce groupe clé de parties prenantes souhaite être impliqué plus en amont dans les initiatives, bénéficier d’une collaboration plus cohérente entre les marchés, ainsi que d’une plus grande transparence et d’une création de valeur partagée renforcée.
Ces attentes soulignent qu’un engagement solide aujourd’hui doit se traduire demain par une collaboration encore plus significative.
Aller vers une co‑création plus approfondie
Au‑delà de la collaboration traditionnelle, une tendance claire se dessine en faveur d’une co‑création plus poussée.
Cela implique notamment :
D’impliquer les organisations de patients plus tôt dans les projets
De co‑concevoir des initiatives de manière plus systématique
De partager davantage les décisions et les responsabilités
Cette évolution est essentielle pour construire des solutions réellement adaptées aux besoins des patients.
Continuer d’écouter et d’évoluer
Nous restons pleinement engagés à écouter, apprendre et adapter nos pratiques afin de renforcer nos partenariats avec les organisations de patients.
En intégrant leurs retours et en favorisant une collaboration toujours plus étroite, nous contribuons à générer un impact positif durable pour les patients et leurs communautés.
« Les retours issus de cette enquête offrent des pistes concrètes pour améliorer nos interactions et renforcer la collaboration. Ils nous aident à mieux comprendre les attentes des organisations de patients et à y répondre de manière plus efficace. »
Aurora Berra
VP Global Patient Office, Ipsen
« Mon rôle est d’être une gardienne de la sécurité des patients. »
En tant que Directrice Safety Science, elle intervient à la fois en développement précoce et en post-commercialisation, veillant à ce que les risques soient anticipés, analysés et maîtrisés pour que les traitements parviennent aux patients en toute responsabilité.
Son travail inclut actuellement un médicament commercialisé et un autre en développement précoce, en préparation pour le premier essai chez l’humain. « Nous devons définir tous les risques potentiels que nous anticipons sur la base des données non cliniques et des composés similaires », explique-t-elle. « Une fois que le premier patient est traité, GPS est très impliqué dans la surveillance de ce qui se passe — chaque événement indésirable signalé, chaque effet secondaire. »
Cette vigilance ne s’arrête pas aux données des essais. « Il s’agit d’analyser et d’interpréter toutes les données de sécurité, qu’elles proviennent des essais cliniques ou de la phase post-commercialisation », dit-elle. « Nous travaillons à identifier toute tendance ou signal pouvant affecter l’équilibre bénéfice–risque des médicaments. Si nous identifions quelque chose, nous décidons des actions à entreprendre. »
Son rôle s’étend également au Benefit Risk Decision Board d’Ipsen. « C’est le plus haut niveau de gouvernance en matière de sécurité », dit-elle. « Les équipes présentent leurs données, et le comité prend les décisions finales. Pour moi, c’est une opportunité unique de comprendre les décisions importantes en matière de sécurité prises pour les produits de l’ensemble du portefeuille Ipsen. »
Un moment marquant a eu lieu lors d’un récent dépôt réglementaire européen pour une extension d’indication. « Une partie de la stratégie consistait à démontrer que le profil de sécurité était cohérent avec ce qui était déjà connu pour le produit », se souvient-elle. « Ce fut un long parcours, mais j’ai tellement appris en chemin, et c’était très gratifiant lorsque la décision finale a été l’approbation. »
Pour Nisha, c’est là tout le sens de la pharmacovigilance. « Comprendre, minimiser et communiquer les risques ou réactions indésirables associés à un médicament, afin de permettre une évaluation du rapport bénéfice–risque », dit-elle. « Ce que nous faisons est crucial pour garantir que les patients puissent accéder à des traitements sûrs. »
Le 18 juin, Ipsen marque la Journée mondiale du cancer du rein (WKCD), un moment mondial pour sensibiliser au cancer du rein et à son impact sur la vie des personnes dans le monde entier.
Le thème de cette année, « Cancer du rein et bien-être émotionnel », met en lumière une réalité importante mais souvent négligée : le cancer du rein n’affecte pas seulement la santé physique des patients.
L’impact caché de la vie avec un CCR
Le cancer du rein est également appelé carcinome à cellules rénales (CCR). Pour de nombreuses personnes vivant avec un CCR, l’impact émotionnel peut être significatif.
Les personnes peuvent ressentir :
De l’anxiété liée aux examens et aux résultats
De l’incertitude quant à l’avenir
Des changements ou des tensions dans les relations personnelles
Un sentiment d’isolement ou de solitude
Une dépression ou une baisse de moral
Ces difficultés peuvent persister tout au long du parcours de la maladie — du diagnostic au traitement, et au-delà. Pourtant, le bien-être émotionnel n’est pas toujours abordé ouvertement dans le cadre des soins courants.
Pourquoi c’est important :
L’écart entre les progrès cliniques et l’expérience vécue reste un défi majeur dans le CCR. Étant donné que de nombreux patients sont diagnostiqués à un stade avancé, des décisions thérapeutiques rapides et efficaces en première ligne peuvent avoir un impact significatif sur les résultats, l’expérience des patients et les options qui s’offriront à eux plus tard dans leur parcours de soins.
Si les avancées thérapeutiques continuent d’améliorer les résultats, les patients et les équipes de soins font encore face à des obstacles importants qui peuvent affecter à la fois la qualité des soins et la qualité de vie. Aider les professionnels de santé à se sentir confiants dans la prise de décisions thérapeutiques opportunes et efficaces en première ligne constitue un levier essentiel pour améliorer l’expérience globale des patients.
L’expérience vécue des patients nous montre que :
Les décisions thérapeutiques au moment du diagnostic sont complexes
L’accès rapide à des traitements efficaces en première ligne est crucial
Le niveau de confiance et d’aisance des professionnels de santé face aux choix thérapeutiques peut varier
Les effets secondaires et leur impact sur la vie quotidienne suscitent des préoccupations
L’inquiétude face à l’évolution de la maladie et aux options futures persiste
Le bien-être émotionnel est donc une composante essentielle d’une prise en charge globale du cancer.
Comment Ipsen soutient la communauté du cancer du rein à l’occasion de cette WKCD
Chez Ipsen, nous nous engageons à soutenir la communauté du cancer du rein — y compris les patients, cliniciens, chercheurs et associations de patients — non seulement en contribuant aux avancées des soins, mais aussi en mettant en avant l’expérience des patients.
À l’occasion de cette Journée mondiale du cancer du rein, Ipsen :
Met en lumière les voix et les expériences des patients
Partage des éclairages sur l’impact émotionnel du CCR
Soutient la sensibilisation aux ressources liées au bien-être émotionnel
Encourage des échanges plus ouverts au sein de la communauté
Par ces actions, nous souhaitons contribuer à une approche des soins plus globale et centrée sur le patient.
Un moment pour réfléchir et agir
La Journée mondiale du cancer du rein est une opportunité pour chacun d’entre nous de réfléchir à l’impact global du cancer.
Nous encourageons les collaborateurs à :
Consulter les contenus WKCD d’Ipsen
Prendre le temps de comprendre l’expérience des patients
Favoriser les échanges autour du bien-être émotionnel dans les soins oncologiques
Placer l’humain au cœur des soins
Alors que nous continuons à façonner l’avenir du CCR, une chose est claire : faire progresser les soins signifie aller au-delà des résultats cliniques afin de mieux prendre en compte l’expérience complète des personnes vivant avec la maladie.
Mariam Soukouna ne s’attendait pas à être touchée. Elle participait à un séminaire interne finance et achats, le genre d’événement centré sur la stratégie, la performance et les priorités opérationnelles. Mais ce jour-là, l’équipe a accueilli une invitée qui n’avait rien à voir avec tout cela : la mère d’un enfant atteint d’une maladie rare.
Elle a parlé de son fils, des années passées à vivre avec une pathologie pour laquelle aucun traitement n’existait, et de ce que représentait le travail d’Ipsen pour leur famille. Un avenir possible. Quelque chose à quoi se raccrocher.
.
Pour Mariam, recevoir ce témoignage n’a pas été anodin. Loin des slides et des tableaux de bord, c’était un rappel direct et sans filtre que derrière chaque décision, chaque contrat, chaque négociation, il y a un patient.
Travailler dans les achats, c’est évoluer à distance des patients et des professionnels de santé. Une distance structurelle, pas intentionnelle. Mais qui peut faire perdre de vue le sens derrière les process. Ce séminaire a comblé cet écart.
Depuis, Mariam porte ce témoignage dans son travail au quotidien. Non pas comme un rappel ponctuel, mais comme quelque chose qui continue d’alimenter son engagement. Parce que comprendre ce à quoi sert vraiment son travail change la façon dont on se lève chaque matin pour le faire.
La cholestase intrahépatique familiale progressive (PFIC) est une maladie complexe. Trouver des informations fiables à son sujet ne devrait pas l’être. PFIC Colors a été conçu pour aider les familles à mieux comprendre cette maladie et à trouver rapidement les informations et le soutien dont elles ont besoin.
La cholestase intrahépatique familiale progressive (PFIC) est un groupe de maladies hépatiques héréditaires rares qui peuvent affecter les personnes de différentes manières. Les symptômes, l’évolution de la maladie et les traitements nécessaires varient selon les différents types de PFIC.1
Cette complexité rend la PFIC difficile à comprendre pour les familles, surtout au début, après le diagnostic. Les familles confrontées à un diagnostic de PFIC décrivent souvent les premiers jours comme accablants, déroutants et isolants. La complexité de la maladie et sa présentation variable d’une personne à l’autre peuvent rendre difficile la compréhension et la recherche d’informations correspondant à leur vécu.
Cette période peut être source de solitude et de confusion, remplie de questions et d’incertitudes. Dans ces moments-là, avoir accès à des ressources fiables, élaborées par la communauté, peut apporter de la clarté et du réconfort face à une situation qui peut paraître insurmontable. C’est précisément ce que les familles peuvent trouver surle site web du Réseau PFIC.
.
Comprendre la PFIC grâce à la couleur
Pour aider les personnes à trouver des informations factuelles, précises et bienveillantes, Ipsen s’est associé à Réseau PFIC afin de développer une campagne positive et engageante diffusée via les canaux numériques, notamment les réseaux sociaux, pour atteindre les personnes recherchant des informations sur la PFIC. L’engagement est encouragé grâce à un contenu qui utilise le concept positif, inspirant et simple de présenter la PFIC comme une maladie aux multiples facettes.
Cette initiative, inspirée par la couleur, vise à mieux comprendre la complexité de la PFIC en orientant les familles vers l’ensemble des ressources disponibles sur le site web du Réseau PFIC. Elle a pour objectif de redonner de la couleur à la vie des personnes concerné·e·s grâce à des informations plus claires et plus accessibles, et à un lien significatif avec une communauté qui comprend leur vécu.
L’importance de la clarté, du lien et de la communauté
Emily Ventura, cofondatrice et directrice générale du Réseau PFIC et mère de Cedar, atteinte de PFIC, a elle-même vécu les difficultés rencontrées lors des premiers stades du diagnostic.
Dans la vidéo ci-dessous, Emily présente plus en détail cette initiative et explique pourquoi il est si important d’apporter clarté, soutien et solidarité aux familles touché·e·s par la PFIC.
« Lorsque ma fille a reçu le diagnostic [de PFIC], en 2012, nous n’avons reçu aucune ressource ni aucun contact avec une quelconque communauté pour obtenir du soutien ; il n’en existait aucune à l’époque. Cela nous a laissé·e·s avec un sentiment de solitude, d’isolement et de peur face à son diagnostic. »
Au fil de ses échanges avec d’autres parents confronté·e·s à des situations similaires, Emily a constaté que de nombreuses personnes atteintes de PFIC se retrouvaient sans accompagnement ni soutien. Ce constat a donné naissance à Réseau PFIC, aujourd’hui une communauté internationale offrant un large éventail d’informations fiables et un réseau de soutien aux personnes touché·e·s par la PFIC et à leurs proches.
.
Explorez les couleurs du PFIC
L’approche PFIC Colors vise à aider les familles à mieux comprendre la PFIC. En organisant l’information en six thèmes, chacun associé à une couleur, et en s’appuyant sur des témoignages concrets, le Réseau PFIC aide les familles à se sentir plus confiantes et plus maîtresses de leur parcours.
Pour en savoir plus sur la PFIC, cliquez sur les encadrés thématiques ci-dessous pour accéder à la section dédiée du site web de Réseau PFIC.
Diagnostic de la PFIC
La PFIC est généralement diagnostiquée chez les nourrissons et les jeunes enfants, mais aussi chez les adolescents et les adultes. Au début, il est normal de ressentir de la peur, de l’isolement et de ne pas savoir vers qui se tourner.
Les symptômes peuvent avoir un impact considérable sur la vie quotidienne, mais ils peuvent être gérés. L’un des plus courants est une démangeaison intense (prurit).
On connaît actuellement 13 types de PFIC, chacun étant causé par des mutations génétiques différentes. Bien que de nombreux symptômes se recoupent entre les différents types de PFIC, chaque type présente des caractéristiques distinctes. De nouveaux types sont régulièrement découverts, ce qui nous permet de mieux comprendre la PFIC et, à terme, d’améliorer la prise en charge.
La PFIC est généralement diagnostiquée chez l’enfant, mais elle peut aussi toucher les adultes, parfois après des années de symptômes hépatiques inexpliqués. Ses manifestations peuvent différer de celles observées chez l’enfant, ce qui peut compliquer le diagnostic. Comprendre le vécu des adultes atteint·e·s de PFIC peut vous aider à trouver le soutien et les soins adaptés.
La PFIC est une maladie héréditaire, c’est-à-dire qu’elle se transmet de génération en génération au sein des familles. Même avec le même type de PFIC, les symptômes peuvent varier considérablement d’une personne à l’autre en fonction des modifications (ou mutations) spécifiques du gène impliqué. De nouvelles mutations sont régulièrement découvertes, ce qui contribue à améliorer le diagnostic, la compréhension et la prise en charge de la PFIC.
Il existe différentes options de traitement, allant des médicaments à la chirurgie en passant par la transplantation hépatique. En discuter avec votre médecin vous permettra de déterminer le traitement le plus adapté à votre situation ou à celle de votre enfant.
Rendez-vous sur PFIC.org pour obtenir toutes les informations concernant le PFIC.
Cette campagne a été développée conjointement par Ipsen et Réseau PFIC.
Par Ivan Diaz-Padilla, Vice-président senior et Responsable mondial de l’Oncologie, Unité Thérapeutique, R&D
La leucémie myéloïde aiguë (LAM) demeure un cancer du sang difficile à traiter. Malgré les progrès réalisés, les rechutes restent fréquentes et le contrôle de la maladie à long terme demeure hors de portée pour de nombreux patients, en particulier les personnes âgées ou celles qui ne peuvent recevoir une chimiothérapie intensive. À mesure que nos connaissances sur la LAM progressent, il apparaît clairement que, pour changer cette réalité, il faut adopter une approche différente.
De l’évasion à l’engagement
La recherche révèle aujourd’hui que la LAM est une maladie complexe et évolutive, dans laquelle le système immunitaire joue un rôle central. En effet, la LAM ne se résume pas à la prolifération incontrôlée de cellules cancéreuses. Elle résulte également d’une interaction constante entre ces cellules et le système immunitaire, interaction qui peut permettre à la maladie d’échapper aux défenses naturelles de l’organisme. Cette nouvelle compréhension transforme notre manière d’envisager le traitement et soulève une question fondamentale : mobiliser et soutenir le système immunitaire du patient pourrait-il ouvrir de nouvelles perspectives thérapeutiques ?
L’idée d’utiliser le système immunitaire pour traiter le cancer n’est pas nouvelle et son efficacité a déjà été démontrée dans certains contextes. Cependant, dans le cas de la LAM, ce type d’approche s’est révélée particulièrement complexe, mettant en lumière la capacité de cette maladie non seulement à échapper au système immunitaire, mais aussi à l’affaiblir activement. Ces observations montrent qu’une activation immunitaire globale ne suffit pas et qu’il est nécessaire de développer des stratégies plus précises et ciblées, conçues pour surmonter les mécanismes de résistance spécifiques à la LAM.
Des stratégies thérapeutiques émergentes
Chez Ipsen, nous faisons progresser cette réflexion en explorant des approches de nouvelle génération visant à mobiliser le système immunitaire. Celles-ci reposent notamment sur l’activation sélective de certaines sous-populations de lymphocytes T, avec un meilleur niveau de précision et de contrôle. Cette stratégie marque une évolution qui s’oriente vers une immunomodulation plus ciblée, ouvrant la voie à une utilisation plus fine et intentionnelle du système immunitaire dans la lutte contre la maladie.
La combinaison de ces nouvelles thérapies ciblant le système immunitaire avec des traitements déjà établis pourrait agir de manière complémentaire. Elle offrirait ainsi une approche prometteuse pour mieux cibler les multiples mécanismes perturbés par la LAM, améliorant ainsi l’efficacité des traitements tout en préservant leur tolérance.
Perspectives
Le besoin de nouvelles approches pour traiter la LAM ne fait aucun doute. Il est tout aussi clair que la prochaine vague de progrès reposera sur une compréhension plus approfondie de la maladie, envisagée comme un système complexe.
Nous pensons que cela implique d’embrasser toute la complexité de la biologie immunitaire et de la traduire en innovations thérapeutiques ciblées et évolutives. Nous ne sous-estimons pas les défis à relever. Les progrès nécessiteront rigueur scientifique, collaboration et persévérance. Mais l’opportunité est réelle : aller au-delà des réponses à court terme pour offrir aux patients qui en ont le plus besoin des bénéfices plus durables.
Philippe Lopes‑Fernandes, Directeur des opérations
La science a évolué – les meilleurs partenariats évoluent avec elle
Il y a dix ans, le développement des médicaments était bien différent. Aujourd’hui, certaines des avancées scientifiques les plus prometteuses émergent de petites équipes très spécialisées, capables d’avancer rapidement — des équipes qui opèrent sous de fortes contraintes opérationnelles et disposent de peu de marge pour tout ce qui pourrait ralentir leur élan.
Or, de nombreuses structures d’accords classiques n’ont pas été conçues pour cette réalité. Elles privilégient la stabilité au moment de la signature : des cadres rigides, fondés sur des hypothèses qui peuvent ne pas résister aux premières données. La science est par essence itérative et imprévisible. Lorsque la structure d’un partenariat — et la manière dont les partenaires collaborent — ne peut évoluer au même rythme, des frictions apparaissent et finissent par nuire aux avancées scientifiques.
Ce que je recherche dans un partenariat va bien au‑delà de la seule structure de l’accord. Il s’agit d’un objectif commun porté par des actions concrètes, d’un lien continu entre les partenaires avant et bien après la signature, et d’un alignement de jugement qui demeure solide, même lorsque la science avance plus vite que ne peut l’anticiper le moindre contrat.
Le défi consiste à bâtir quelque chose d’assez robuste pour durer, tout en restant suffisamment flexible pour s’adapter. Les meilleures structures d’accords ne sont pas des solutions standard. Elles sont conçues spécifiquement pour le partenariat en question — et c’est là que la créativité, l’innovation et une volonté constante d’évoluer prennent tout leur sens.
Construire des structures d’accords créatives
Aucun partenaire ne se ressemble ; aucun accord ne devrait donc être identique. Notre point de départ est toujours une compréhension sincère : non seulement de ce que chaque partenaire apporte, mais aussi de ce qui permettra réellement à la collaboration de fonctionner pour lui. Où nos forces complémentaires peuvent‑elles accélérer la science de la manière la plus efficace ? Quels sont les moments clés de décision ? À quoi ressemble le succès à chaque étape du développement scientifique ?
La structure de l’accord est le résultat de ce dialogue — et non l’application d’un modèle prédéfini. C’est ce qui en fait une démarche créative : une conception sur mesure, pensée pour servir une science ambitieuse et transformatrice.
La preuve dans le pipeline
Depuis 2020, Ipsen a construit plus de 35 programmes grâce à des partenariats. En 2025 seulement, trois accords dont je suis particulièrement fier — chacun structuré différemment, chacun façonné autour d’une science et d’un partenaire spécifiques — sont unis par un objectif commun : faire émerger une science audacieuse, capable de transformer les paradigmes thérapeutiques.
L’avenir appartient à des structures d’accords flexibles, personnalisées et adaptées à la réalité actuelle du développement des médicaments : des équipes agiles, des cycles rapides et des contraintes bien réelles. C’est le niveau d’exigence que nous nous fixons, et notre pipeline en est le reflet
En mobilisant les bons partenaires tout au long de notre chaîne de valeur et en impliquant nos collaborateurs pour avançer ensemble
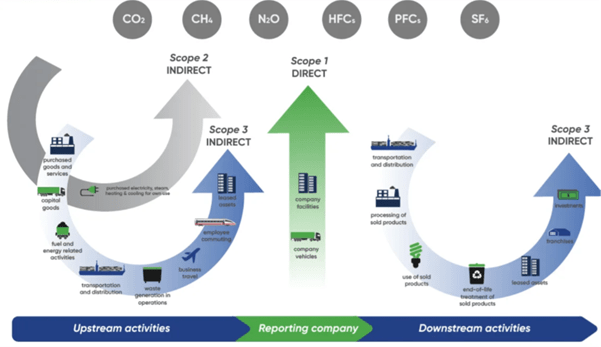
Pour une entreprise comme Ipsen, l’impact environnemental ne se limite pas à ses opérations. En réalité, cela ne représente qu’une petite partie du tableau.
La majeure partie de notre empreinte se situe ailleurs, tout au long de notre chaîne de valeur. Elle est déterminée par les partenaires avec lesquels nous travaillons, les matières premières que nous utilisons, ainsi que la manière dont nos médicaments sont développés et distribués. Aujourd’hui, environ 90 % de nos émissions relèvent du Scope 3, ce qui reflète la complexité de l’écosystème pharmaceutique.
Cette réalité redéfinit notre approche du développement durable.
Source: From https://ghgprotocol.org/.
Ces dernières années, Ipsen a réalisé des progrès significatifs dans la réduction des émissions liées à ses propres activités. Les émissions de scope 1 et 2 ont été réduites de plus de moitié par rapport à 2019, grâce à des améliorations en matière d’efficacité énergétique, d’électrification et de choix opérationnels plus durables. En parallèle, nous avons également pris des mesures concernant des éléments clés de notre empreinte de scope 3, notamment les voyages d’affaires, la flotte de véhicules, les emballages et les trajets domicile-travail des employés, des domaines où le changement peut être mis en œuvre plus directement. Ces résultats démontrent que des progrès sont déjà en cours et que la croissance et la responsabilité environnementale peuvent aller de pair.
La prochaine étape est différente. Il s’agit moins de ce que nous pouvons faire seuls que de ce que nous pouvons accomplir ensemble.
La prochaine étape est différente. Il s’agit moins de ce que nous pouvons faire seuls que de ce que nous pouvons accomplir ensemble.
Nous étendons désormais notre approche au Scope 3, passant des ambitions et des cadres conceptuels à la mise en œuvre concrète avec nos fournisseurs et partenaires. La réduction de ces émissions nécessite un changement dans le mode de fonctionnement de tout un écosystème. Cela implique de collaborer avec nos principaux fournisseurs pour mieux comprendre leurs émissions et identifier les domaines où des réductions significatives peuvent être réalisées. Cela implique de passer des estimations à des données plus précises, permettant ainsi des actions ciblées et mesurables. Et cela signifie intégrer la durabilité dans les processus métier fondamentaux de notre chaîne d’approvisionnement, de la stratégie aux opérations.
Dans un secteur hautement réglementé comme celui des produits pharmaceutiques, le changement ne se fait pas du jour au lendemain. Les chaînes d’approvisionnement sont complexes, et même des évolutions en apparence simples, comme l’emballage, nécessitent du temps, une validation et une autorisation réglementaire. Mais ces contraintes ne limitent pas notre ambition. Elles définissent la voie à suivre.
Chez Ipsen, nous pensons que le progrès repose sur une responsabilité partagée. Il se construit grâce à des partenariats à long terme, à une amélioration continue et à la volonté de s’attaquer aux aspects les plus complexes du défi.
À l’occasion de la Journée mondiale de l’environnement, le message est clair : nous voulons réduire notre impact et nous agissons là où cela compte le plus. En nous appuyant sur ce que nous avons déjà accompli, nous accélérons notre action tout au long de notre chaîne de valeur.
Par Christelle Huguet, EVP & Directrice R&D, Ipsen
En recherche biopharmaceutique, tous les défis ne se valent pas. Certaines des opportunités les plus porteuses se trouvent dans des domaines marqués par une biologie complexe, des données limitées et des patients qui attendent depuis bien trop longtemps des options thérapeutiques efficaces.
Pourquoi nous choisissons d’agir là où d’autres hésitent
Chez Ipsen, nous avons fait le choix délibéré de nous concentrer sur les besoins médicaux les plus élevés. Ces domaines exigent une compréhension scientifique approfondie, une grande capacité d’adaptation et un engagement sur le long terme. Mais ils offrent aussi le plus fort potentiel pour générer un impact véritable pour les patients.
Les maladies aux besoins médicaux non satisfaits se situent souvent en dehors des modèles de développement traditionnels et nécessitent de nouvelles façons de penser et de travailler. Les populations de patients peuvent être restreintes ou hétérogènes. Les critères d’évaluation peuvent être plus difficiles à définir. La biologie peut encore être en cours d’élucidation. C’est pourtant précisément dans ces contextes que l’innovation peut être la plus impactante et que les avancées peuvent changer des vies.
Transformer l’ambition scientifique en progrès concrets
Relever ces défis implique d’accepter l’incertitude. Cela nous pousse à poser de meilleures questions, à renforcer la collaboration et à concevoir des programmes de développement plus intelligents.
Nous développons une compréhension approfondie des mécanismes des maladies et nous adaptons en permanence notre approche à mesure que de nouvelles connaissances émergent. Cela nous permet de concevoir des programmes à la fois scientifiquement rigoureux et étroitement alignés aux besoins des patients. En appliquant cette discipline, nous pouvons prendre des risques réfléchis et intentionnels, en privilégiant des programmes ayant un potentiel firstinclass ou bestinclass et en ouvrant de nouvelles perspectives là où il en existait peu auparavant.
Placer l’impact au cœur des attentes
En nous attaquant aux défis les plus complexes, notre ambition n’est pas seulement de développer de nouveaux traitements, mais aussi de contribuer à redéfinir la manière dont le progrès est mesuré dans des populations complexes et rares. Ce faisant, nous cherchons à élever les attentes et à démontrer qu’une innovation rigoureuse et centrée sur le patient est possible.
S’attaquer aux problématiques les plus complexes n’est pas le chemin le plus aisé. Mais pour les patients présentant des besoins médicaux non satisfaits, c’est le chemin essentiel, et celui que nous choisissons avec fierté, en faveur du progrès scientifique.
Par Sandra Silvestri, pour Ipsen.com
Dans un environnement thérapeutique en constante évolution, l’autorisation d’un médicament ne constitue plus la fin du parcours: ce n’est que le début. Les données en vie réelle (real‑world evidence, RWE), les analyses post‑hoc et les études de phase IV structurées ouvrent de nouvelles perspectives pour l’apprentissage scientifique. Chez Ipsen, nous nous engageons à explorer les questions encore sans réponse et à produire des données robustes qui vont au‑delà du développement initial du médicament, afin d’apporter une vision plus large et à long terme, fondée sur des preuves, au service des communautés que nous accompagnons.
Le rôle croissant des données en vie réelle
Les essais cliniques apportent des réponses scientifiques essentielles, mais par nature, ils ne peuvent pas refléter pleinement la réalité du quotidien des patients : la diversité des parcours, la complexité des maladies et l’imprévisibilité des prises en charge.
Les données en vie réelle contribuent à combler cet écart. Elles rendent compte de ce qui compte réellement pour les personnes concernées : l’efficacité d’un traitement sur la durée, sa tolérance au quotidien et sa place dans la vie réelle avec la maladie. En investissant dans des programmes solides de RWE, nous approfondissons notre compréhension des besoins des patients, mesurons l’impact réel de nos médicaments et veillons à ce qu’ils continuent d’apporter des bénéfices concrets aux personnes qui en dépendent.
Analyses post‑hoc et essais de phaseIV: approfondir la compréhension là c’est essentiel
Les analyses post‑hoc et les études de phase IV — réalisées après l’autorisation d’un médicament — jouent un rôle tout aussi fondamental. En tenant compte de la diversité des patients et la variabilité des parcours de soins, nous questionnons des hypothèses afin d’en tirer des enseignements adaptés aux défis du monde réel. Quel est l’impact de nos médicaments chez des patients présentant certaines comorbidités ou recevant d’autres traitements ? Quels marqueurs cliniques pourraient mériter une exploration approfondie ? Comment l’environnement thérapeutique, et par conséquent les recommandations de traitement, évoluent‑ils au fil du temps ?
En adoptant une approche ciblée, nous concevons des analyses capables de répondre aux questions les plus importantes pour les professionnels de santé comme pour les patients, contribuant ainsi à une meilleure compréhension de la maladie et à une prise en charge toujours plus individualisée.
Un avenir fondé sur l’apprentissage continu
Les enseignements issus des données en vie réelle, combinés à la richesse des essais de phase IV et des analyses post‑hoc, offrent une compréhension multidimensionnelle et approfondie de l’impact d’un médicament. Pour Ipsen, la génération en continue de données après l’autorisation de mise sur le marché n’est pas un simple complément : c’est un engagement. Un engagement à comprendre pleinement l’impact de nos médicaments, non seulement pour le plus grand nombre, mais aussi pour chaque individu, avec ses particularités, qui mérite des réponses à ses questions.
L’avenir de la prise en charge des patients reposera sur cette culture de l’apprentissage continu. L’autorisation d’un médicament est une étape importante, qui mérite d’être célébrée, mais elle ne marque pas la fin du parcours. Chaque donnée issue du monde réel, chaque résultat à long terme, chaque analyse post‑autorisation nous rapproche d’une optimisation des soins pour les patients qui en ont le plus besoin.
Notre ambition est claire : faire en sorte que le progrès scientifique ne s’arrête pas à l’autorisation, mais qu’il s’accélère grâce à elle — afin que chaque patient bénéficie non seulement d’une innovation thérapeutique, qui continue d’évoluer grâce à de nouvelles données, une approche ciblée et une raison d’être.
Vous allez maintenant quitter le site web global du groupe Ipsen. Pour continuer, veuillez cliquer sur Continuer ?
Nous apprécions vos commentaires
Avez-vous trouvé qu'il était facile de naviguer sur le site web d'Ipsen et de trouver les informations que vous recherchiez ?